Overview of Trancription and Chromatin Regulation
p>It is well established that nucleosomes play fundamentally important roles
in the organization and maintenance of the genome. Nucleosome modifications
have been shown to be associated with transcriptional regulation at well-studied
genes and models have emerged that connect regulation of gene expression to
histone modification by specific chromatin regulators. We have carried out a
systematic genome-wide analysis of nucleosome acetylation and methylation at
sufficient resolution to determine whether models that connect regulation of
gene expression to histone modification apply to gene regulation throughout
the yeast genome.
The results described here, taken together with recent discoveries, are consistent
with the following general model connecting gene expression to histone modification.
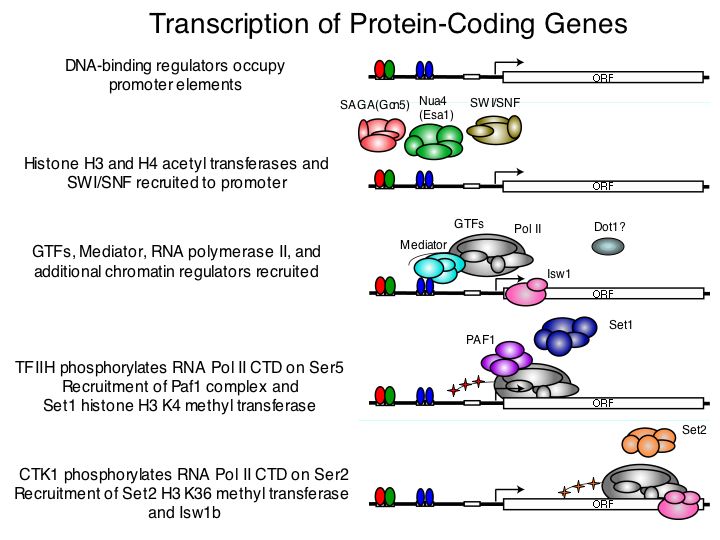
Transcriptional activation by DNA binding regulators generally involves recruitment
of Gcn5 and Esa1 to promoters, where these HATs acetylate specific residues
on histones H3 and H4 at local nucleosomes. We were able to find few exceptions
to this general rule, where only one or the other HAT acetylates its target
residues at the promoters of actively transcribed genes. Active transcription
is characteristically accompanied by histone H3K4 trimethylation by Set1 at
the beginning of genes, and by H3K4 dimethylation and monomethylation at nucleosomes
positioned further downstream in the transcription unit. As the transcription
apparatus proceeds down the transcription unit, increasing levels of histone
H3K36 trimethylation are observed at most active genes, catalyzed by Set2. Histone
H3K79me3, which is catalyzed by Dot1, is enriched within genes, but unlike the
other modifications studied here, this enrichment is not clearly associated
with active transcription.
The genome-wide maps of histone occupancy and modification described here should
provide investigators with information useful for further exploring the histone
code and its implications for gene regulation and chromosome organization and
maintenance. We expect that the approaches used here to map histone occupancy
and modification in yeast can also be used to gain insights into the linkage
between gene expression and histone modification across the genome in higher
eukaryotes.